Tutorials: Learn How to Use DBTMEE
CASE 1: Identification of Fertilization-specific Genes (Not Expressed in Parthenogenetic Development)
CASE 2: Inference of the TFs that Potentially Bind to Genes Expressed after Fertilization
CASE 3: Inferring the Effect of Histone Modifications in Sperm on Early Embryo Development
CASE 4: Identification of Potential Factors that Enhance Reprogramming to Induced Pluripotent Stem Cells
CASE 3: Inferring the Effect of Histone Modifications in Sperm on Early Embryo Development
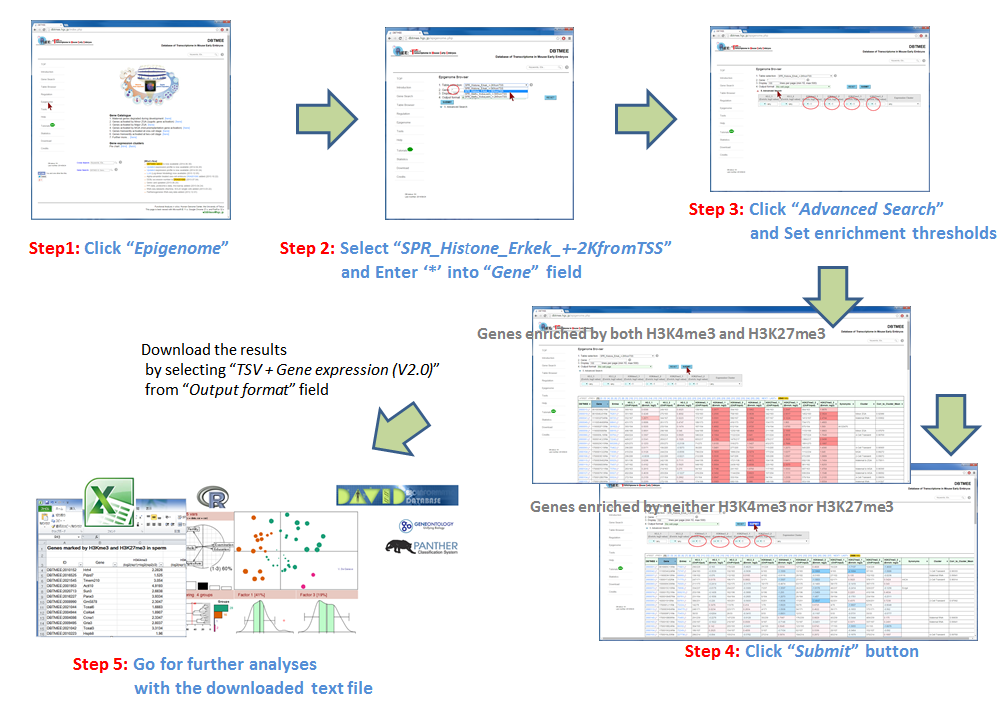
Question: How does epigenetic inheritance from sperm affect zygotic gene expression? The Epigenome Browser provides epigenetic data, such as sequencing-based DNA methylations in sperm and histone modifications in sperm. The "Advanced Search" option performs an 'AND' search operator with the given thresholds. As an example, we entered an asterisk (*, a wildcard that matches all gene names) into the Gene field, then selected "SPR_Histone_Erkek_+-2KfromTSS" from the Table selection field (Step 2). We have set the advanced search option with >1.0 for H3K4me_1, H3K4me_2, H3K27me_1, and H3K27me_2 (Step 3). This setting implies that gene promoters (+-2kbp from TSS) are marked by all four ChIP replications of the histones in sperm (i.e. 2-fold enrichment in ChIP against input). This returns 3,543 genes (Step 4-1). In addition, the setting of the advanced search option with <=1.0 for the four ChIP experiments (i.e. marked by neither histone modifications) returns 3,686 genes (Step 4-2). User can download these results as text file by selecting "TSV+Gene expression (V2.0)" from the Output format field, then can use them for further analyses (Step 5). Since this output format provides FPKMs and expression patterns also, for example, users can analyze whether fertilization-specific genes (see CASE 1 also) are marked by those histone modifications in sperm; after gathering genes that were not expressed at both p1C and p4C (<=1.0 in FPKMs) but were expressed at least one stage (>1.0 in FPKMs), we could prepare 658 and 901 genes that were marked by both histone modifications and by neither histone modifications, respectively. The distribution of FPKMs of these genes along the stages and cell types suggests that after fertilization the bivalent genes categorized into 4-cell transient, minor ZGA, and MGA patterns tend to be actively transcribed. In particular, the increases of gene expression levels are remarkable at ESC and MEF. This result might be helpful to identify genes that are affected by sperm transmission and to determine the roles of epigenetic inheritance from sperm (Example). |
[Step 1] [Step 2] 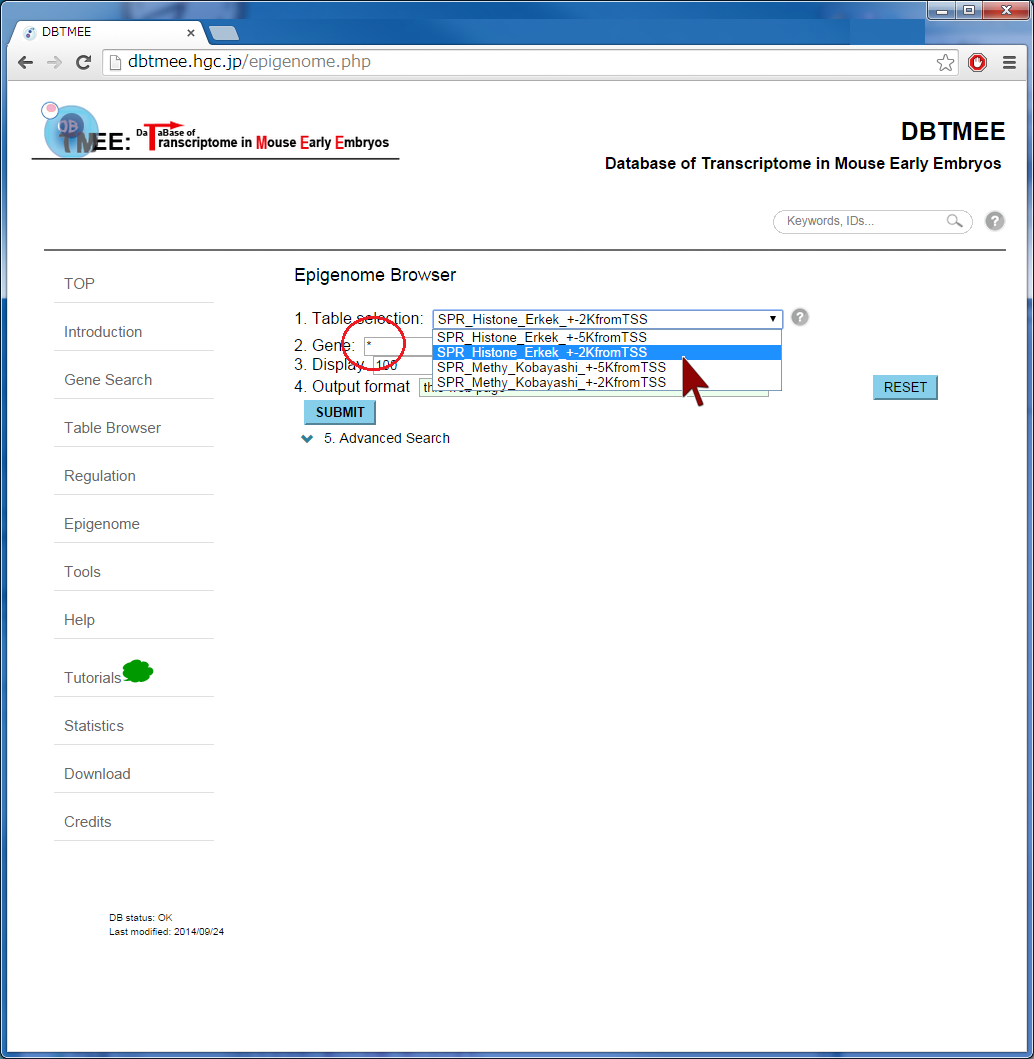 [Step 3] 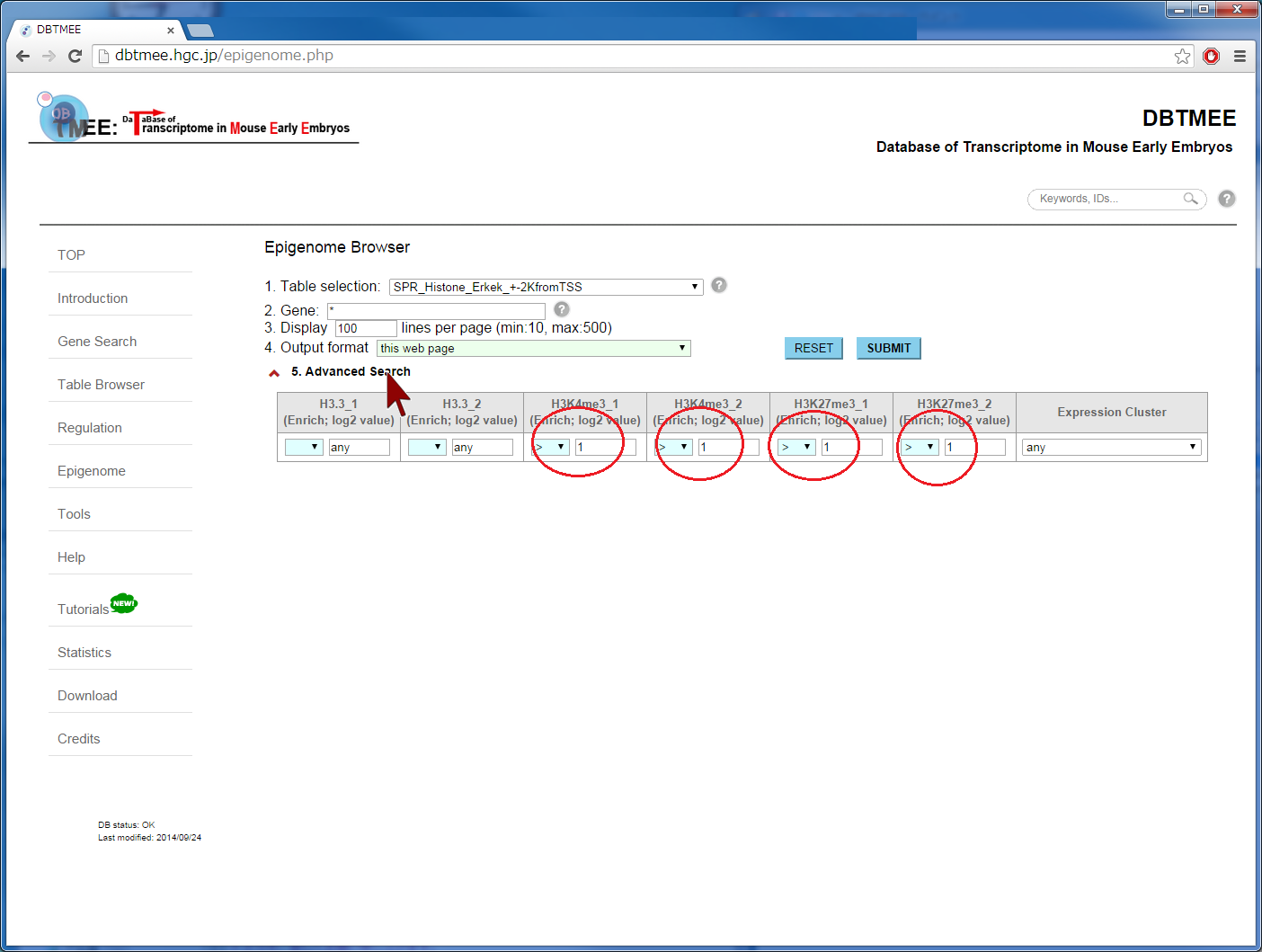 [Step 4-1: bivalent genes] 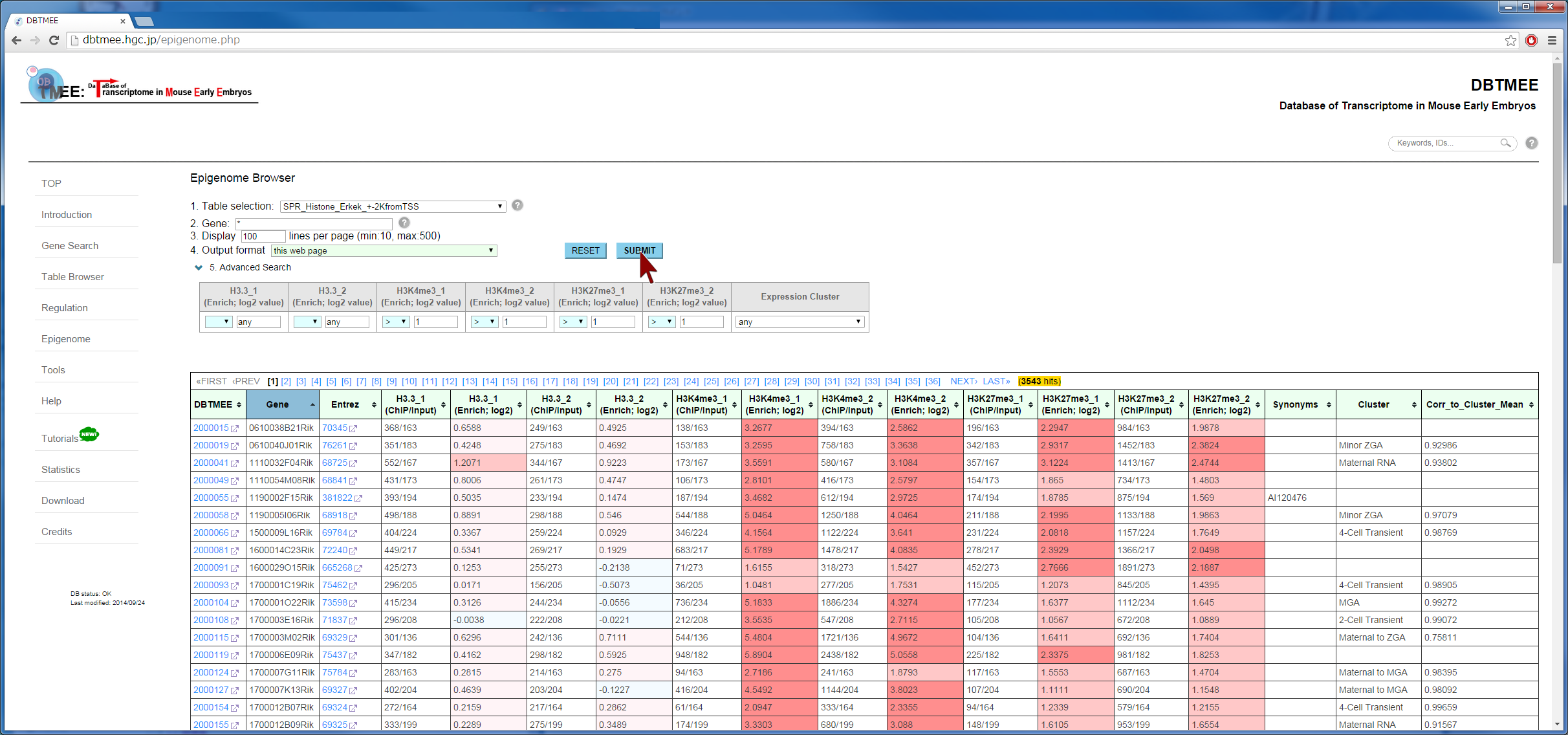 [Step 4-2: histone-free genes]  [Step 5]  [Example of further analysis] 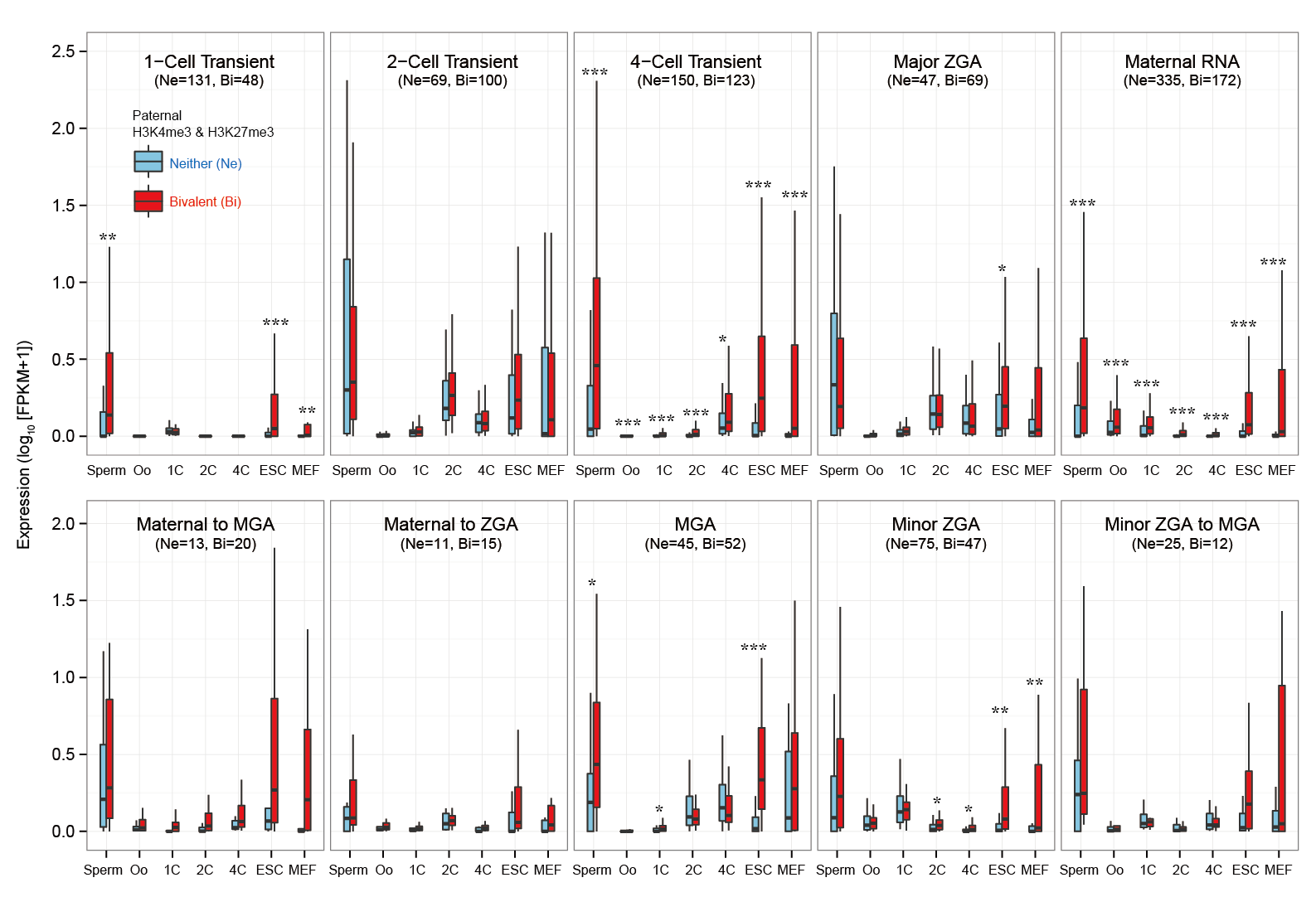 |

{kind=link}
{kind=link}